
近日,上海交通大學環(huán)境科學與工程學院大氣污染控制與模擬團隊馬磊副教授在《Nature Communications》上發(fā)表了題為“Temperature-driven mechanistic transition in propylene oxidation over Pt/CeO2 ensemble catalysts”的研究成果。該研究揭示了Pt/CeO2團簇催化劑在丙烯(C3H6)氧化反應中溫度驅(qū)動的動態(tài)機制轉(zhuǎn)變,通過氫活化策略顯著提升了催化氧化性能,明確了金屬Pt團簇為本征催化活性位點,闡明了170 °C閾值溫度時反應路徑發(fā)生根本變化。這不僅為高效貴金屬催化劑設計提供了新思路,還對機動車尾氣凈化等實際應用具有重要指導意義。論文第一完成單位為上海交通大學環(huán)境科學與工程學院綠色造紙與資源循環(huán)全國重點實驗室。
研究簡介
C3H6作為活性較高的烯烴類揮發(fā)性有機物(VOCs),在大氣光化學過程中對臭氧(O3)生成具有重要作用,同時其氧化產(chǎn)物也可貢獻一定量的二次有機氣溶膠(SOA)。丙烯催化氧化是機動車尾氣污染控制中的關鍵反應之一,Pt/CeO2催化劑因其高活性和氧存儲能力而備受關注。然而,長期以來,Pt/CeO2催化劑的反應活性位點以及溫度對反應機制的影響存在爭議。本研究通過簡單的氫活化處理,成功構建了金屬Pt團簇活性位點,使50%丙烯氧化轉(zhuǎn)化率的反應溫度(T50)降低了120 °C。研究進一步確認了丙烯氧化的動態(tài)催化反應路徑,即低溫下雙分子吸附,高溫下界面活化主導。結(jié)合多種原位表征技術和理論計算,研究發(fā)現(xiàn)反應路徑在170 °C附近存在一個閾值溫度:低溫時丙烯氧化遵循經(jīng)典的Langmuir-Hinshelwood模型,而高溫時O2活化位點從Pt團簇表面遷移到Pt-O-Ce界面,這一動態(tài)轉(zhuǎn)變機制為理解催化劑在真實工況下的催化反應行為提供了新視角。
研究內(nèi)容
催化反應性能提升,氫活化的關鍵作用:研究通過氫活化處理Pt/CeO2催化劑(Pte-300A),顯著提升了丙烯氧化活性。如圖1所示,未活化的Pte催化劑T50約為282 °C,而Pte-300A的T50降至160 °C,表觀活化能從138.0 kJ/mol降低至111.5 kJ/mol。對比其他Pt基催化劑,Pte-300A表現(xiàn)出優(yōu)異的反應速率,且在水蒸氣存在下穩(wěn)定性良好,具備實際應用潛力。

圖1. 負載型Pt催化劑上C3H6氧化的催化活性與表觀活化能。a C3H6氧化起燃曲線。反應條件:1000 ppm C3H6,10% O2,N2平衡氣,質(zhì)量空速(WHSV)為240,000 mL g?1 h?1;b C3H6氧化的阿倫紐斯曲線;c Pte-300A催化劑與文獻報道的Pt催化劑在C3H6氧化反應活性的對比。
催化劑結(jié)構分析表征,金屬Pt團簇活性位點:通過高角度環(huán)形暗場掃描透射電子顯微鏡(HAADF-STEM)和X射線吸收光譜(XAS)等技術,揭示了氫活化引起的結(jié)構重構。如圖2所示,未活化的Pte催化劑以單層Pt團簇為主(直徑0.45 nm),而Pte-300A形成了多層Pt團簇(直徑0.84 nm),團簇頂層為金屬Pt位點(Pt0)。原位DRIFTS和XPS結(jié)果進一步證實,Pt0位點具有更高的電子密度和吸附反應特性,是丙烯氧化的關鍵活性位點。

圖2. Pte和Pte-300A催化劑的結(jié)構表征與活性位點確認。(a,b)Pte和Pte-300A的HAADF-STEM圖像(黃色圓圈:Pt單原子;紅色虛線圓圈:平面單層Pt團簇;紅色實線圓圈:多層Pt團簇);(c)Pte和Pte-300A在30 ℃下CO吸附的原位DRIFTS光譜;(d)Pte和Pte-300A的歸一化Pt L3邊XANES譜;(e)Pte和Pte-300A在R空間中的傅里葉變換k2加權EXAFS譜;(f)Pt箔、PtO2、Pte和Pte-300A樣品的Pt L3邊EXAFS小波變換圖。
界面性質(zhì)動態(tài)變化,170 °C閾值溫度的發(fā)現(xiàn):利用原位拉曼光譜(In situ Raman)、近常壓XPS(NAP-XPS)和電子能量損失譜(EELS),追蹤了反應過程中催化劑界面的演化。如圖3所示,當溫度低于170 °C時,CeO2的氧空位和過氧化物物種濃度穩(wěn)定。一旦溫度超過170 °C,氧空位被填充,Ce3+比例下降,表明O2在Pt-O-Ce界面活化并參與反應。這一閾值溫度在CO催化氧化中也得到驗證,相關理論具有普適性。

圖3. Pte-300A催化劑在C3H6氧化過程中的動態(tài)變化。(a)Pte-300A催化劑上100至250 ℃溫度區(qū)間內(nèi)C3H6氧化的原位拉曼光譜;(b)100至225 ℃溫度范圍內(nèi)過氧化物比例與缺陷峰強度比(ID/IF2g)的變化趨勢;(c)不同溫度下Ce 3d軌道的近常壓X射線光電子能譜(NAP-XPS);(d)和(e)分別為Pte-300A在162 ℃和188 ℃反應后的電子能量損失譜(EELS)分析結(jié)果。
溫度驅(qū)動反應機制變化,從Langmuir-Hinshelwood到界面活化:通過原位DRIFTS和動力學研究,闡明了反應路徑的溫度依賴性。如圖4所示,低溫時(如162 °C),C3H6和O2競爭吸附在金屬Pt團簇上,遵循Langmuir-Hinshelwood模型,氧輔助的sp3 C-H鍵脫氫為速率決定步驟。高溫時(如188 °C),C3H6完全覆蓋Pt位點,O2則在Pt-O-Ce界面活化,兩者表面覆蓋度發(fā)生顯著變化,反應級數(shù)變化表明機制轉(zhuǎn)變。DFT計算驗證了Pt0位點具有更低的脫氫能壘(1.53 eV vs. Ptδ+的2.41 eV),支持了實驗結(jié)論。研究通過多尺度原位反應表征與理論計算結(jié)合,揭示了溫度驅(qū)動下催化劑界面和反應機制的動態(tài)演變過程,這一發(fā)現(xiàn)不僅深化了對催化反應構效關系的理解,還為優(yōu)化設計負載型貴金屬催化劑提供了理論依據(jù)。
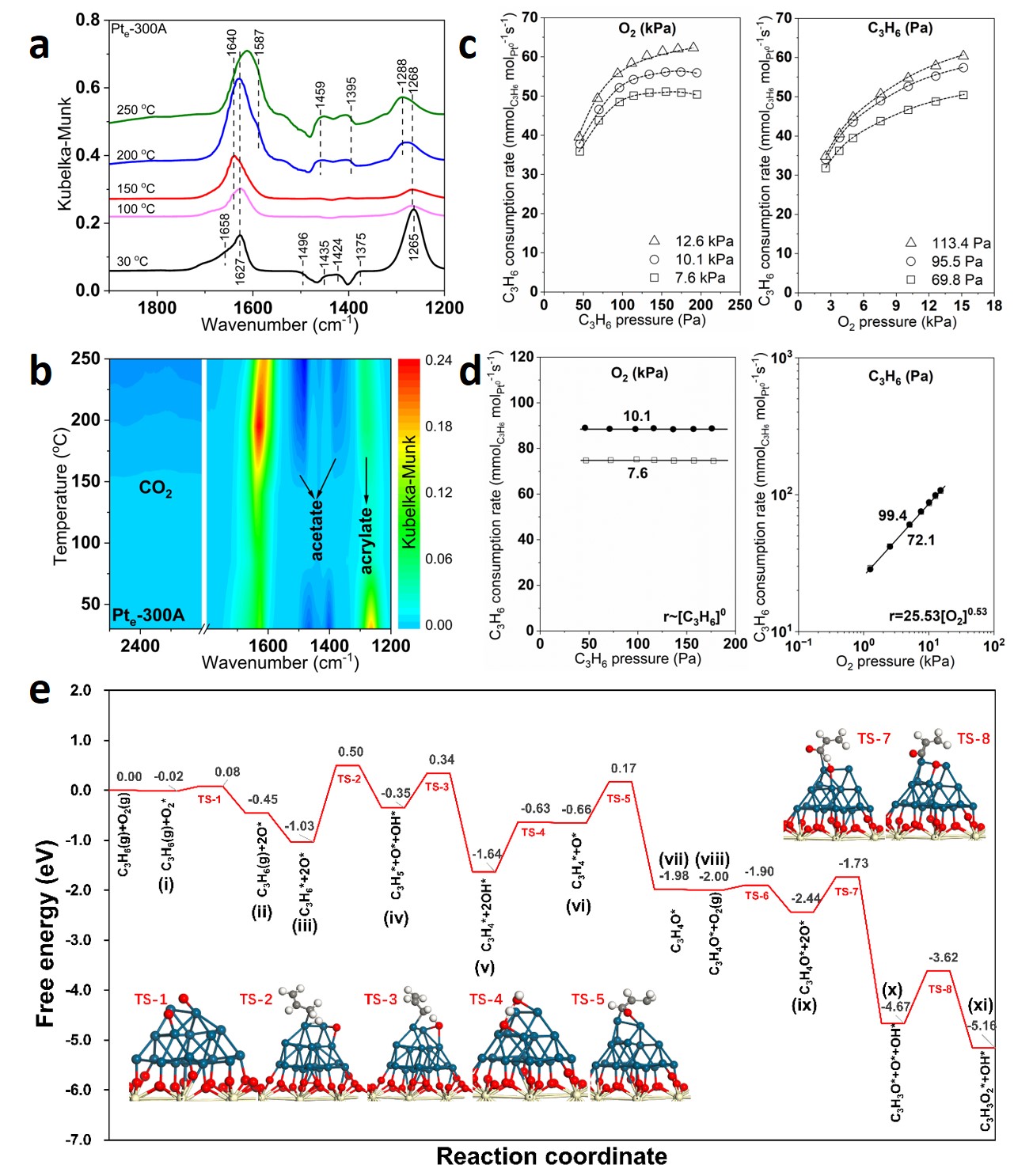
圖4. Pte-300A催化劑上C3H6氧化反應機理與表面中間體的研究。(a)C3H6與O2穩(wěn)態(tài)共吸附反應的原位DRIFTS光譜;(b)C3H6氧化的二維等高線圖;(c)和(d)分別展示了162 ℃和188 ℃條件下C3H6與O2分壓對Pte-300A催化劑上C3H6消耗速率的影響;(e)DFT計算獲得的C3H6氧化反應機理與反應能壘,包括詳細過渡態(tài)和自由能變化。
作者簡介
論文第一作者為上海交通大學環(huán)境科學與工程學院2024屆博士畢業(yè)生李梓豪(現(xiàn)就職于中國石油集團石油化工研究院),論文通訊作者為上海交通大學馬磊副教授、加州大學河濱分校劉福東副教授,合作單位包括華東理工大學、中國人民大學、移動源污染排放控制技術國家工程實驗室等。